AMBER é uma família de campos de força para mecânica molecular frequentemente utilizada para modelagem de biomoléculas, como proteínas e sequęncias de DNA. Desenvolvido pelo grupo do pesquisador Peter Kollman, da Universidade da Califórnia, o pacote recebe o mesmo nome do software de mecânica molecular que originalmente simula esse campo de força. Atualmente, entretanto, os parâmetros desse campo podem ser aplicados em outros softwares alternativos, como o Tinker.
Como descrito no artigo sobre campos de força, cada campo de força segue uma funçăo particular para a descriçăo de cada um dos termos da equaçăo geral de potencial. Dentro da família de campos de força do AMBER, a forma funcional mante-se, entretanto, para cada campo de força particular, um grupo de parâmetros distintos será atribuído.
A energia potencial dos campos AMBER obedece a seguinte funçăo:
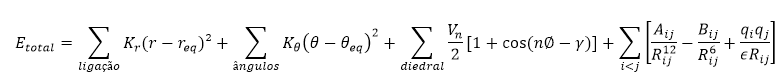
As tręs primeiras somatórias săo referentes ŕ energia potencial da ligaçăo covalente, enquanto o último termo é referente ŕs interaçőes năo-covalentes (eletrostáticas e de Van der Walls).
Os significados de cada termo săo:
• Somatória da ligaçăo: representa a energia entre dois átomos covalentemente ligados. A ligaçăo é considerada uma mola, e por esse motivo, é descrita pela funçăo harmônica da mola ideal. O termo Kr é referente ŕ constante de mola da ligaçăo, há também os termos da distância da ligaçăo e a distância de equilíbrio (parâmetro do campo).
• Somatória do ângulo: representa a energia angular entre tręs átomos covalentemente ligados. Seguindo o mesmo princípio do item citado acima, a energia será referente ŕ perturbaçăo do ângulo considerado comparado ao ângulo de equilíbrio do trio de átomos.
• Somatória diedral: representa a energia da torçăo de uma ligaçăo sobre sua ordem e as ligaçőes das redondezas ou pares isolados de elétrons.
• Último termo: o ultimo termo representa as forças năo covalentes atuantes sobre o sistema, como as interaçőes eletrostáticas e as interaçőes de Van der Walls. Os dois primeiros termos dentro dessa somatória săo referentes ŕs interaçőes de Van der Walls, que no caso dessa família de campos de força, utiliza o potencial de Lennard-Jones. O último termo, por sua vez, descreve as interaçőes eletrostáticas descritas pelas leis de Coulomb.
A parametrizaçăo dos campos depende do ambiente modelado pelo campo e do tipo de molécula a ser considerado. Sendo a mecânica molecular completamente baseada em parâmetros, toda e qualquer propriedade que năo pode ser descrita pela forma funcional é colocada como uma alteraçăo no parâmetro, de forma que os dados obtidos sejam coerentes com o experimental.
Sendo assim, um campo desenvolvido para a modelagem de fase gasosa terá parâmetros diferentes de um campo desenvolvido para a modelagem em fase aquosa, uma vez que todo o potencial de solvataçăo deve ser descrito nos parâmetros dos átomos.
No caso de tipos de moléculas diferentes, temos, por exemplo, um campo desenvolvido para macromoléculas (proteínas e cadeias nucleotídicas) contra um campo desenvolvido para moléculas orgânicas pequenas. Em macromoléculas, existem forças que podem ser ignoradas – como a força de dipolo – devido ao tamanho da molécula, visto que numa somatória final, essas forças acabarăo se anulando. Em moléculas orgânicas pequenas, entretanto, é necessário que exista maior refinamento nos parâmetros, para que a descriçăo seja a mais acurada possível.
O AMBER possui algumas séries de campos:
• Série ff (ff94, ff96, ff98, ff99): utilizado para peptídeos, proteínas e cadeias de nucleotídeos;
• GAFF: utilizado para pequenas moléculas orgânicas;
• GLYCAM: utilizado para carboidratos;
[1] J. Wang, P. Cieplak and P. A. Kollman, "How Well Does a Restrained
Electrostatic Potential (RESP) Model Perform in Calcluating Conformational
Energies of Organic and Biological Molecules?", J. Comput. Chem., 21,
1049-1074 (2000)
[2] W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr.,
D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman,
"A Second Generation Force Field for the Simulation of Proteins, Nucleic
Acids, and Organic Molecules", J. Am. Chem. Soc., 117, 5179-5197 (1995)